|
1
|
|
|
2
|
- Predicting the binding affinity between a target and a ligand forming a
non-covalent complex is a very challenging task that has been pursued
for decades.
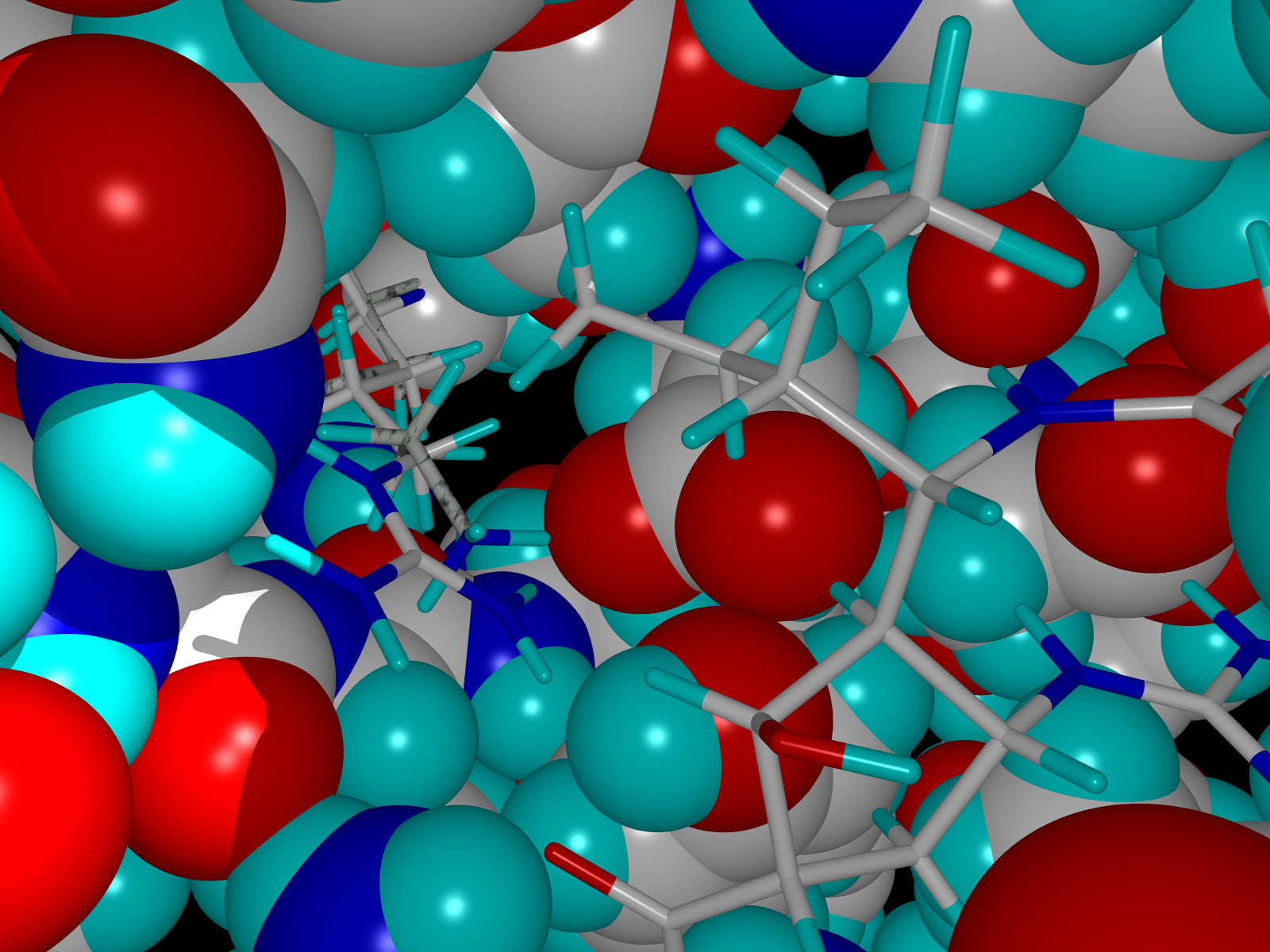
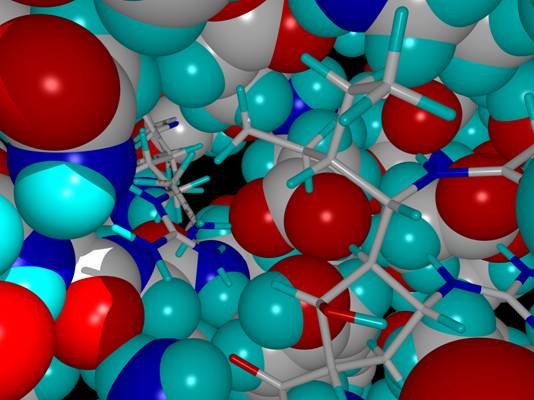
- Computer-aided structure-based methods are aimed at predicting the
binding mode of a ligand in the binding site of a protein or any
molecular target and at obtaining an estimate of the binding affinity.
- These methods involve two computational steps: docking and scoring.
- .
|
|
3
|
- In the docking step, multiple protein-ligand configurations are
generated.
- Then, a scoring function is used to calculate the affinity between the
receptor and the ligand for each configuration
- Requirements for the scoring function:
- 1) Binding free energies for each configuration need to be estimated
accurately
- 2) A scoring function must be sufficiently fast to be applied in a
docking algorithm.
|
|
4
|
|
|
5
|
|
|
6
|
|
|
7
|
|
|
8
|
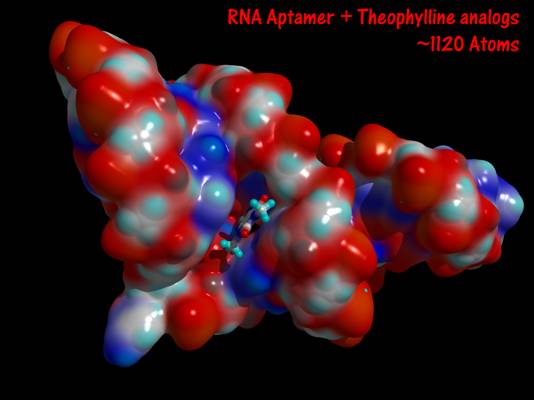
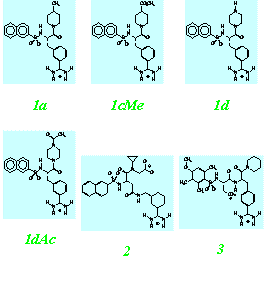
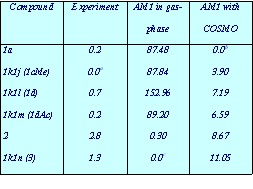
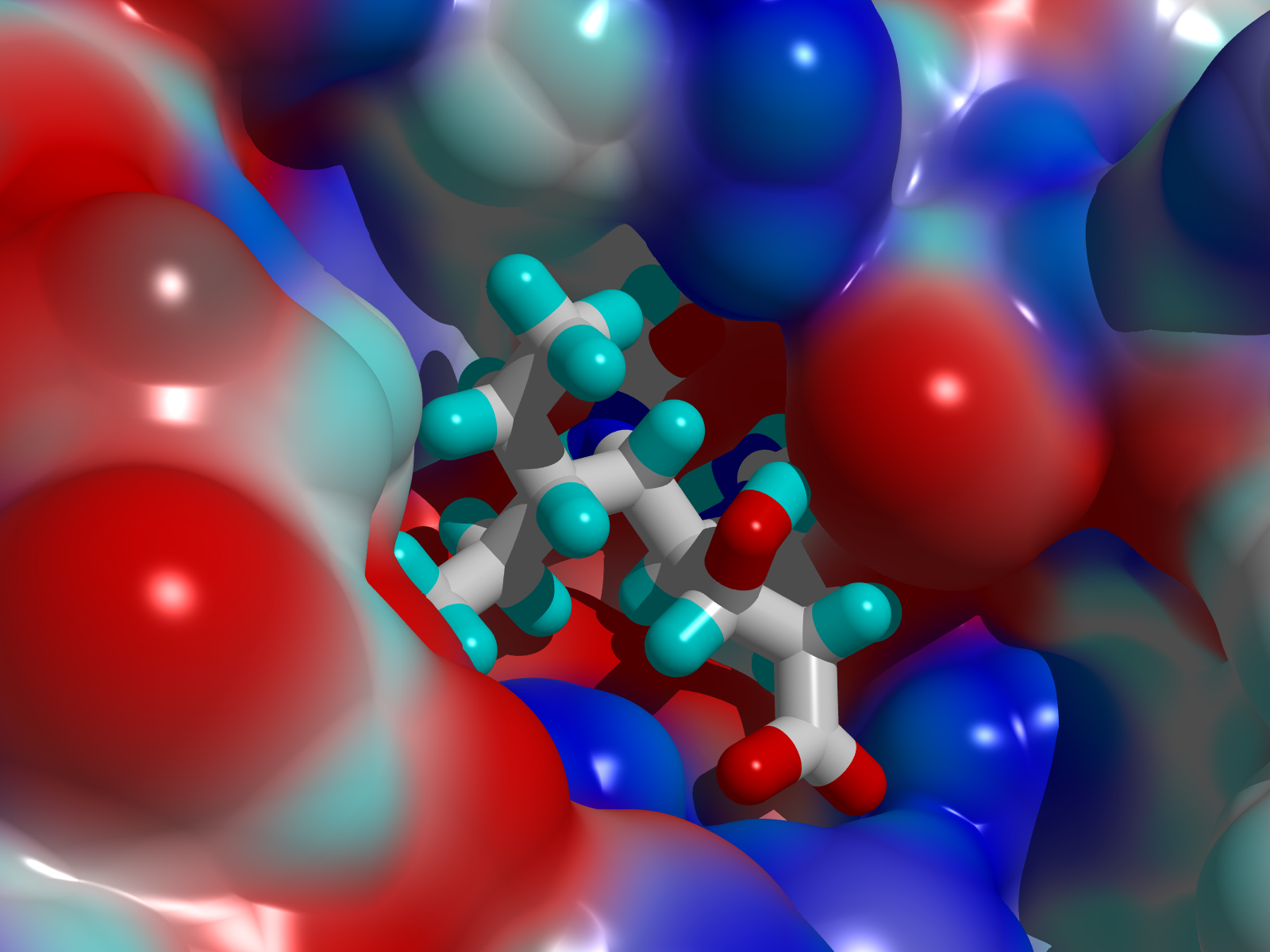
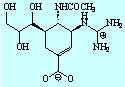
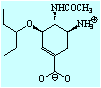
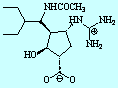
- Theophylline-binding RNA aptamer
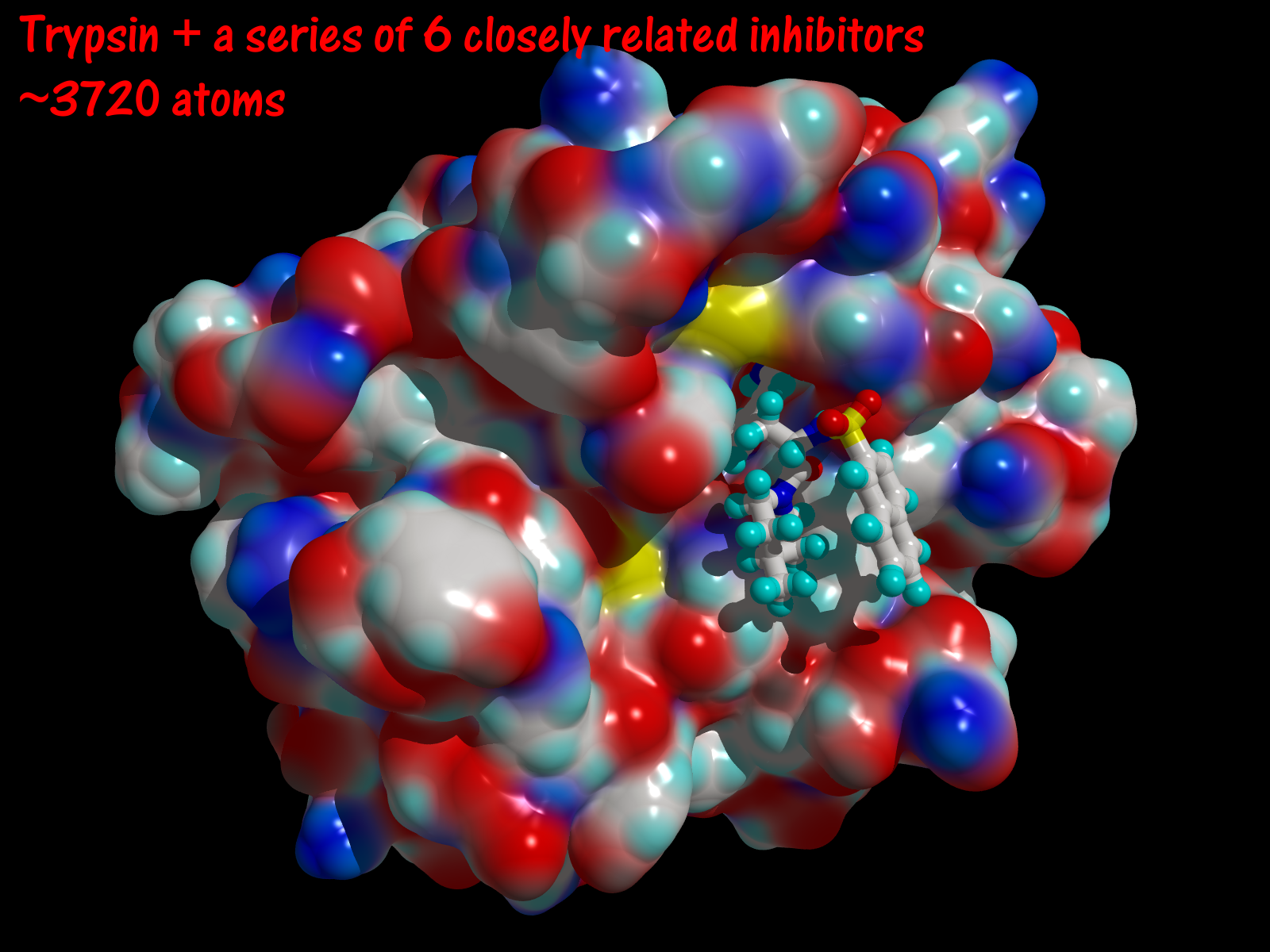
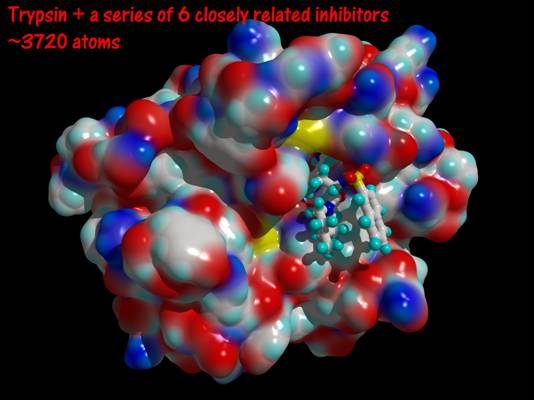
- Trypsin and a series of closely related inhibitors
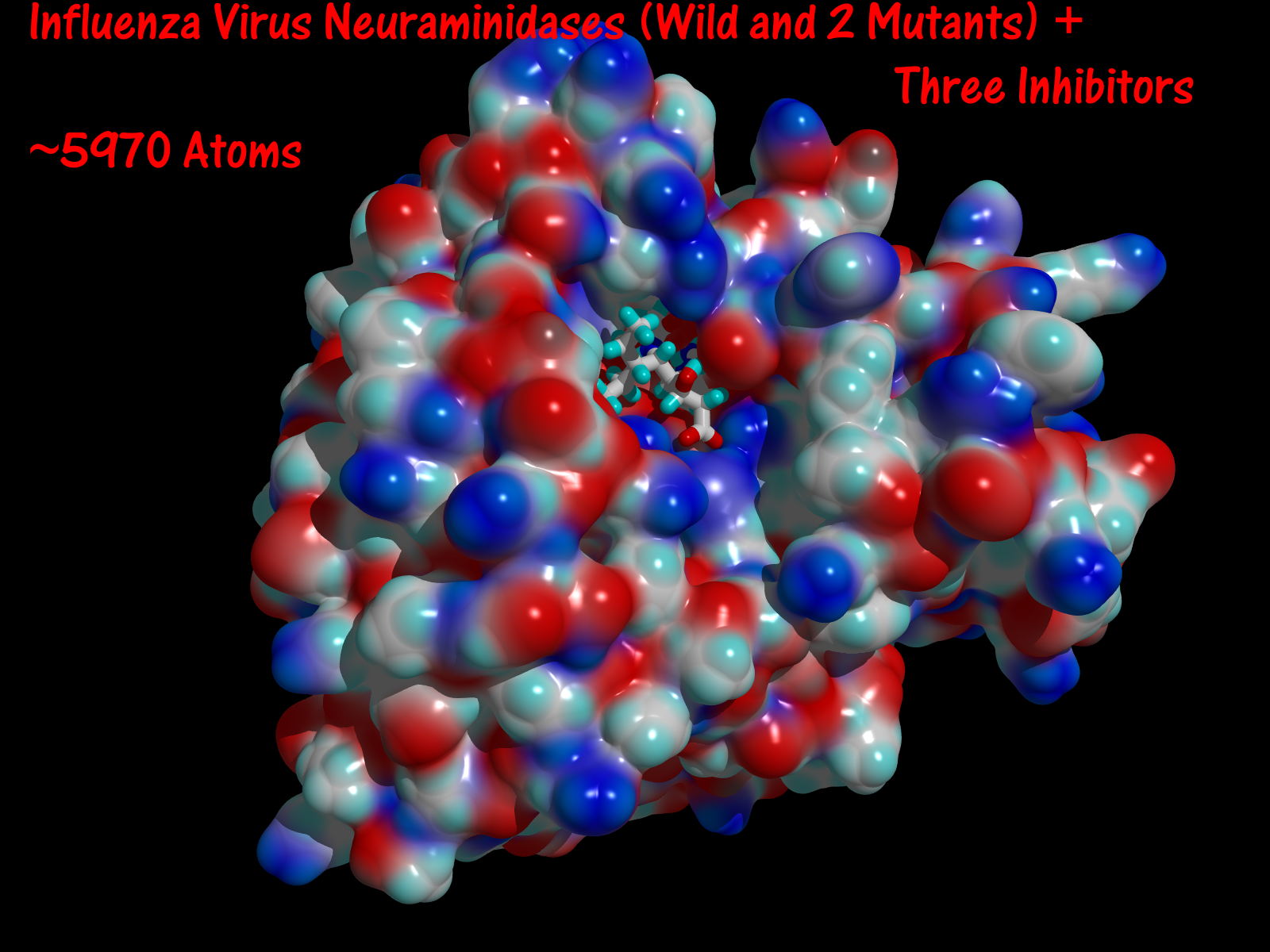
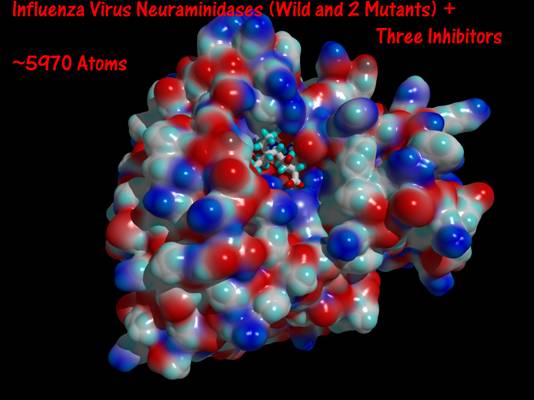
- Influenza virus neuramindase (wild + mutants) and its three inhibitors
|
|
9
|
|
|
10
|
|
|
11
|
|
|
12
|
|
|
13
|
|
|
14
|
|
|
15
|
|
|
16
|
|
|
17
|
|
|
18
|
|
|
19
|
|
 Notes
Notes{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}