Objective
The aim of this tutorial is estimation of protein-ligand binding energy in non-covalent complex between the 1cgh and a small ligand using MM-PBSA approach.
The acronym MM-PBSA stands for Molecular Mechanics- Poisson Bolzmann Surface Area
The MM-PBSA approach represents the postprocessing method to evaluate free energies of binding or to calculate absolute free energies of molecules in solution
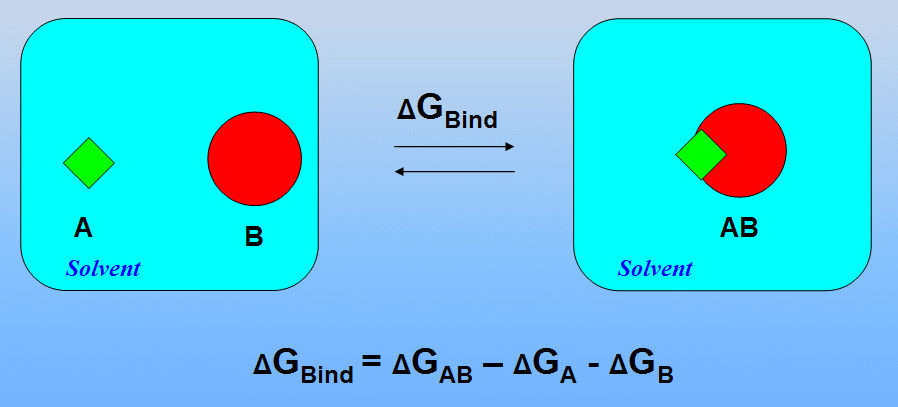
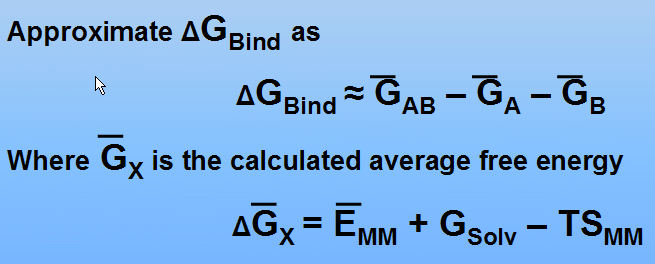
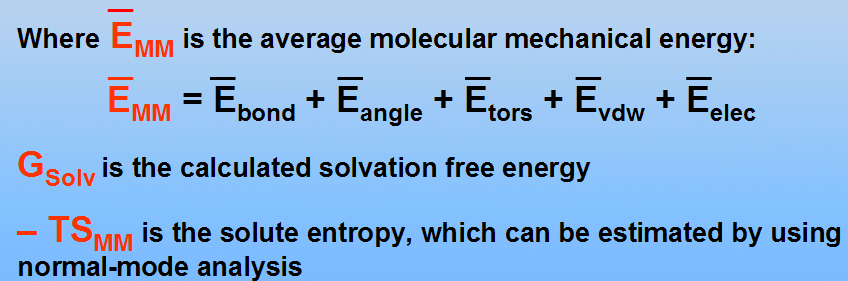
(see P.A. Kollman at al., Calculating Structures and Free Energies of Complex Molecules: Combining Molecular Mechanics and Continuum Models, Acc. Chem. Res. 2000, 33, 889-897 for details)
In this tutorial we shall omit calculation of entropy contributions (-TSMM term)
We shall carry out three independent MD simulations of the complex and both individual proteins in a periodic box with solvent saving a set of representative structures:
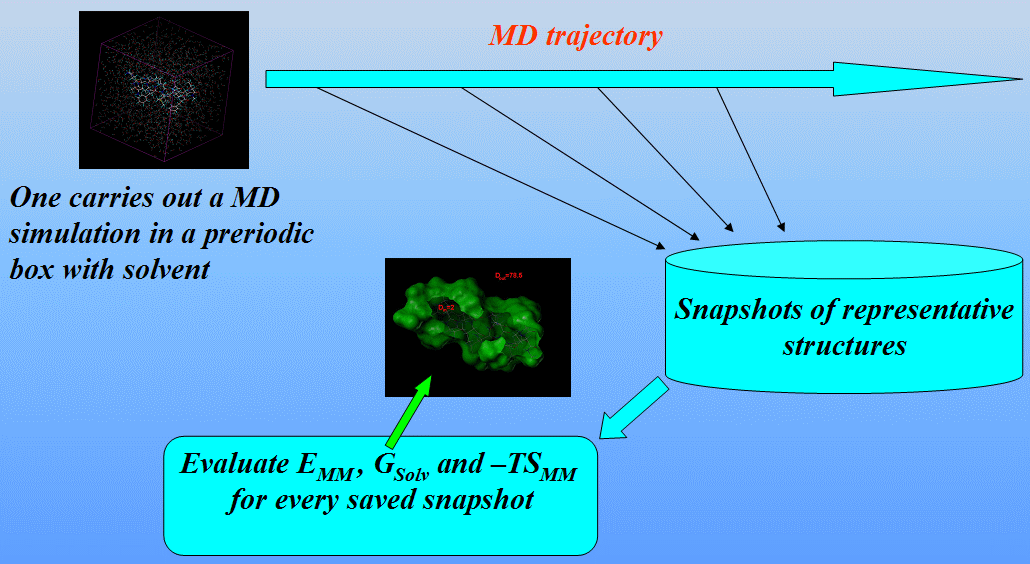
We shall use the mm_pbsa.pl script from the AMBER9 distribution to automatically perform all the necessary steps to estimate the binding free energy.
So, this tutorial consists of the following steps:
2) Obtain equilibrated systems using sander
3) Run the production simulation and save representative snapshots using sander
4) Postprocess obtained snapshots removing all solvent molecules using mm_pbsa.pl script
5) Calculate the binding free energy using mm_pbsa.pl script
Step 1: Preparation of the necessary topology and coordinate files for the complex and both individual proteins in a periodic box with solvent using Xleap
We shall use in Xleap structures prepared for the previous tutorial. First, we shall prepare copies of the complex, protein and ligand:
ligand-wat = copy ligand
protein-wat = copy protein
complex-wat=copy complex
Solvate them and save corresponding topology and coordinate files:
solvateoct ligand-wat TIP3PBOX 12
solvateoct protein-wat TIP3PBOX 12
solvateoct complex-wat TIP3PBOX 12
saveamberparm ligand-wat ligand-wat.top ligand-wat.crd
saveamberparm protein-wat protein-wat.top protein-wat.crd
saveamberparm complex-wat complex-wat.top complex-wat.crd
Also, save our systems as PDB files for visual inspection:
savepdb ligand-wat ligand-wat.pdb
savepdb protein-wat protein-wat.pdb
savepdb complex-wat complex-wat.pdb
Step 2: Obtain equilibrated systems using sander
We shall equilibrate the solvated systems by carrying out short minimisations (1000 steps) and 10ps of heating and constant pressure equilibration at 300K. All simulations will be run with shake on hydrogen atoms, a 2 fs time step and langevin dynamics for temperature control. The input files are as follows.
Minimize systems for 1000 steps:
$AMBERHOME/exe/sander -O -i opt-all.in -o ligand-wat-all-minimize.out \
-c ligand-wat.crd -p ligand-wat.top -r ligand-wat-all-minimize.rst
$AMBERHOME/exe/sander -O -i opt-all.in -o protein-wat-all-minimize.out \
-c protein-wat.crd -p protein-wat.top -r protein-wat-all-minimize.rst
$AMBERHOME/exe/sander -O -i opt-all.in -o complex-wat-all-minimize.out \
-c complex-wat.crd -p complex-wat.top -r complex-wat-all-minimize.rst
Equilibrate systems for 10 ps:
$AMBERHOME/exe/sander -O -i equilibrate.in -o ligand-wat-equilib.out -c \
ligand-wat-all-minimize.rst -p ligand-wat.top -r ligand-wat-equilib.rst
$AMBERHOME/exe/sander -O -i equilibrate.in -o protein-wat-equilib.out \
-c protein-wat-all-minimize.rst -p protein-wat.top -r protein-wat-equilib.rst
$AMBERHOME/exe/sander -O -i equilibrate.in -o complex-wat-equilib.out \
-c complex-wat-all-minimize.rst -p complex-wat.top -r complex-wat-equilib.rst
In this tutorial we use relatively short equilibration but in general case be sure to proceed with the MM-PBSA production MD run only in the case if the systems have equilibrated (temperature, density, total energy, RMSD, etc.).
Step 3: Run the production simulation and save representative snapshots using sander
We shall run a total of 50 ps saving snapshots every 1 ps. Note that for our systems simulation time of 50 ps is most likely too short to obtain a set of representative snapshots that adequately sample the equilibrium ensemble. A value of several ns or so would probably be more appropriate. However, this will suffice for the purposes of this tutorial.
$AMBERHOME/exe/sander -O -i production.in -o ligand-wat-prod.out -c ligand-wat-equilib.rst \
-p ligand-wat.top -r ligand-wat-prod.rst -x ligand-wat-prod.mdcrd
$AMBERHOME/exe/sander -O -i production.in -o protein-wat-prod.out -c protein-wat-equilib.rst \
-p protein-wat.top -r protein-wat-prod.rst -x protein-wat-prod.mdcrd
$AMBERHOME/exe/sander -O -i production.in -o complex-wat-prod.out -c complex-wat-equilib.rst \
-p complex-wat.top -r complex-wat-prod.rst -x complex-wat-prod.mdcrd
Step 4: Postprocess obtained snapshots removing all solvent molecules using mm_pbsa.pl script
The general procedure is to edit the mm_pbsa.in file, and then to run the code as follows:
mm_pbsa.pl mm_pbsa.in > mm_pbsa.log
The mm_pbsa.in file refers to "receptor", "ligand" and "complex", but the chemical nature of these is up to the user, and these could equally well be referred to as "A", "B", and "AB". The procedure can also be used to estimate the free energy of a single species, and this is usually considered to be the "receptor".
The output files are labeled ".out", and the most useful summaries are in the "statistics.out" files. These give averages and standard deviations for various quantities, using the following labeling scheme:
First, we shall postprocess a trajectory for the ligand;
mkdir GenerateSnapshots
cd GenerateSnapshots
mkdir LIGAND
cd LIGAND
$AMBERHOME/exe/mm_pbsa.pl ligand-snapshots.in > ligand_coords.log
The meaning of some options in the file ligand-snapshots.in is following:
@GENERAL
PREFIX 1CGH
PATH ./
COMPLEX 1
RECEPTOR 1
LIGAND 1
COMPT ../../complex.top - location of topology files
RECPT ../../protein.top
LIGPT ../../ligand.top
GC 1 - Snapshots are generated from trajectories
AS 0
DC 0
MM 0
GB 0
PB 0
MS 0
NM 0
@MAKECRD
BOX YES - trajectory files contain periodic box information
NTOTAL 5784 - Total number of atoms per snapshot
NSTART 1 - Start structure extraction
NSTOP 50 - Stop structure extraction
NFREQ 1 - Every NFREQ structure will be extracted
#
NUMBER_LIG_GROUPS 1
LSTART 1 - Number of first ligand atom in the trajectory
LSTOP 54 - Number of last ligand atom in the trajectory
NUMBER_REC_GROUPS 1
RSTART 55 - Number of first receptor atom in the trajectory
RSTOP 55 - Number of last receptor atom in the trajectory
@TRAJECTORY
TRAJECTORY ../../ligand-wat-prod.mdcrd
mm_pbsa.pl script extracts coordinates for complex, ligand and receptor. Since our trajectory file contains coordinates of ligand and solvent only and does not contain coordinates of the "complex" and "receptor", we set values RSTART and RSTOP just to comply with input rules of mm_pbsa.pl script.
Since we don't need extracted coordinates of the "complex" and "receptor" we can safely delete them:
rm 1CGH_com.* 1CGH_rec.*
and move ligand's snapshots into the parent directory:
mv 1CGH* ..
Now we shall perform similar procedure for the complex and protein trajectories:
cd ..
mkdir PROTEIN
cd PROTEIN
$AMBERHOME/exe/mm_pbsa.pl protein-snapshots.in > protein_coords.log
rm 1CGH_com* 1CGH_lig*
mv 1CGH* ..
cd ..
mkdir COMPLEX
cd COMPLEX
$AMBERHOME/exe/mm_pbsa.pl complex-snapshots.in > complex_coords.log
rm 1CGH_lig* 1CGH_rec*
mv 1CGH* ..
cd ..
Now directory GenerateSnapshots contains files: 1CGH_lig.crd.xx, 1CGH_rec.crd.xx, and 1CGH_com.crd.xx which correspond to processed snapshots of ligand, protein and their complex, correspondingly.
Step 5: Calculate the binding free energy using mm_pbsa.pl script
After extraction of the snapshots we shall now calculate the interaction energies and solvation free energies for complex, receptor and ligand and average the results to obtain an estimate of the binding free energy.
We will carry out the binding energy calculation using both the MM-PBSA and the MM-GBSA method for comparison. We shall use mm_pbsa.pl script and correspondingly modified input file:
$AMBERHOME/exe/mm_pbsa.pl binding-energy.in > binding-energy.log
The meaning of some options in the file binding-energy.in is following:
PREFIX 1CGH PATH ./ COMPLEX 1 RECEPTOR 1 LIGAND 1 # COMPT ../complex.top - location of topology files RECPT ../protein.top LIGPT ../ligand.top # GC 0 AS 0 DC 0 # MM 1 - Calculation of gas phase energies using sander GB 1 - Calculation of desolvation free energies using the GB models in sander PB 1 - Calculation of desolvation free energies using numerical solutions of the PB equation MS 1 - Calculation of nonpolar contributions to desolvation # NM 0 # @PB PROC 2 - the pbsa program of the AMBER suite is used for solving the PB equation REFE 0 INDI 1.0 - Dielectric constant for the solute EXDI 80.0 - Dielectric constant for the surrounding solvent SCALE 2 LINIT 1000 PRBRAD 1.4 ISTRNG 0.0 RADIOPT 0 NPOPT 1 CAVITY_SURFTEN 0.0072 CAVITY_OFFSET 0.00 # SURFTEN 0.0072 SURFOFF 0.00 # @MM DIELC 1.0 - Dielectricity constant for electrostatic interactions # @GB IGB 2 - Onufriev's GB GBSA 1 SALTCON 0.00 EXTDIEL 80.0 INTDIEL 1.0 # SURFTEN 0.0072 SURFOFF 0.00 @MS PROBE 0.0
The various portions of this input file specify which calculations to run. On which files to run them and any special parameters necessary to calculate the different contributions to the binding free energy. File binding-energy.in contains explanations of the different terms. Values for the different parameters are determined based on empirical data and are subject to on-going research. Refer to publications in the field for more information.
The mm_pbsa.pl script produces several output files, 1CGH_com.all.out , 1CGH_lig.all.out, 1CGH_rec.all.out, and 1CGH_statistics.out along with the binding-energy.log
The log file, just shows if the calculation completed successfully. The *all.out files give the individual energy contributions for each of the snapshots for each of the species.binding-energy.log,
The most important file for us is 1CGH_statistics.out It contains the final averaged binding free energy results:
# COMPLEX RECEPTOR LIGAND # ----------------------- ----------------------- ----------------------- # MEAN STD MEAN STD MEAN STD # ======================= ======================= ======================= ELE -10405.24 286.72 -10418.95 314.94 -5.69 1.98 VDW 1536.44 424.99 1602.62 374.71 4.85 2.10 INT 4749.32 44.32 4659.55 44.19 71.65 5.48 GAS -4119.49 463.17 -4156.78 418.76 70.81 5.11 PBSUR 95.61 6.21 95.04 6.91 4.84 0.06 PBCAL -3370.98 294.71 -3259.27 324.90 -100.82 2.12 PBSOL -3275.37 288.57 -3164.22 318.06 -95.98 2.11 PBELE -13776.22 33.84 -13678.22 39.64 -106.51 1.24 PBTOT -7394.86 431.39 -7321.00 379.12 -25.16 5.09 GBSUR 95.61 6.21 95.04 6.91 4.84 0.06 GB -3624.15 281.72 -3507.66 318.05 -103.58 2.26 GBSOL -3528.54 275.59 -3412.61 311.25 -98.74 2.25 GBELE -14029.39 25.18 -13926.61 26.87 -109.27 1.37 GBTOT -7648.03 429.32 -7569.39 380.22 -27.93 5.23
# DELTA
# -----------------------
# MEAN STD
# =======================
ELE 19.40 95.95
VDW -71.03 544.09
INT 18.11 64.43
GAS -33.52 544.50
PBSUR -4.27 1.51
PBCAL -10.90 81.56
PBSOL -15.17 80.60
PBELE 8.50 46.36
PBTOT -48.69 547.16
GBSUR -4.27 1.51
GB -12.91 85.46
GBSOL -17.19 84.60
GBELE 6.49 33.34
GBTOT -50.71 547.53