A Hybrid QM/MM Simulation
This tutorial explains how to set up QM/MM simulation using AMBER 9
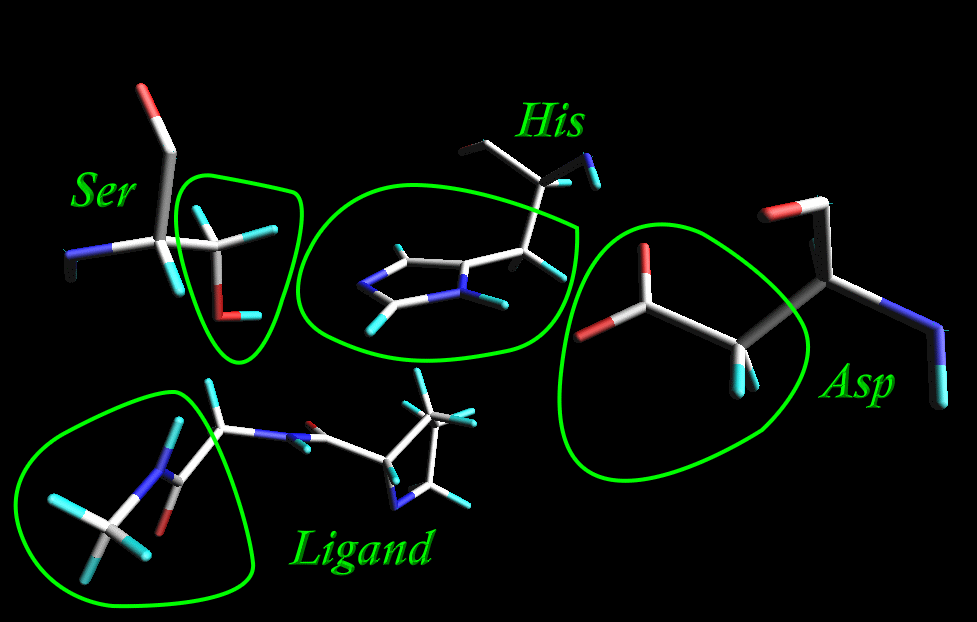
We are going to treat "catalytic triad", Ser-His-Asp, as well as part of a ligand qunatum chemically while the rest of a protein Molecular Mechanically.
We shall follow a simple rule for cutting covalent bonds between QM and MM regions:

Ser: 2935, 2936, 2937, 2938, 2939
His: 668, 669, 670, 671, 672, 673, 674,675, 676, 677, 678
Asp: 1394-1399
Ligand: 3668-3675
Our control input file for sander :
Example QMMM Script for Sander 9 &cntrl imin=1, maxcyc=500, (Perform minimization for 500 steps) ntb=0, (Non-periodic simulation) cut=20., (20 angstrom classical non-bond cut off) ifqnt=1 (Switch on QM/MM coupled potential) &end &qmmm qmmask='@668-678,1394-1399,2935-2939,3668-3675' (Treat these atoms using QM) qmcharge=-1, (Charge on QM region is -1) qmtheory=1, (Use the PM3 semi-empirical Hamiltonian) qmcut=20.0 (Use 20 angstrom cut off for QM region) &end
$AMBERHOME/exe/sander -O -i qm-min.in \ -o qm-min.out -c complex.crd \ -p complex.top -r complex-min.rst <dummy
Lets take a look at the output file, qm-min.out.
After cutting out QM part from the protein the total charge on the MM part is not an integral number any more. However, sander automatically adjust it:
QMMM: ADJUSTING CHARGES
QMMM: ----------------------------------------------------------------------
QMMM: adjust_q = 2
QMMM: Uniformally adjusting the charge of MM atoms to conserve total charge.
QMMM: qm_charge = -1
QMMM: QM atom RESP charge sum (inc MM link) = -0.590
QMMM: Adjusting each MM atom resp charge by = 0.000
QMMM: Sum of MM + QM region is now = 0.000
QMMM: ----------------------------------------------------------------------
Then sander informs us about about covalent bonds between QM and MM parts which should be cut:
QMMM: Link Atom Information
QMMM: ------------------------------------------------------------------------
QMMM: nlink = 4 Link Coords Resp Charges
QMMM: MM(typ) - QM(typ) X Y Z MM QM
QMMM: 666 CT 668 CT 15.901 38.980 12.135 0.119 -0.123
QMMM: 1392 CT 1394 CT 21.612 39.103 9.903 0.007 -0.048
QMMM: 2933 CT 2935 CT 14.674 33.952 6.090 0.118 0.147
QMMM: 3665 CT 3668 C 12.591 37.051 4.554 -0.129 0.581
QMMM: ------------------------------------------------------------------------
Then sander prints out coordinates of the QM atom including automatically generated protons:
QMMM: QM Region Cartesian Coordinates (*=link atom)
QMMM: NO. ATOM X Y Z
QMMM: 1 C 15.7580 39.5420 11.2120
QMMM: 2 H 15.0797 40.3883 11.3204
QMMM: 3 H 16.7522 39.9050 10.9513
...
QMMM: 28 H 14.4723 34.1200 3.6299
QMMM: 29 H 13.1032 33.0885 4.1079
QMMM: 30 H 13.2225 33.6912 2.4377
QMMM: 31 *H 15.9015 38.9800 12.1349
QMMM: 32 *H 21.6124 39.1029 9.9025
QMMM: 33 *H 14.6738 33.9521 6.0897
QMMM: 34 *H 12.5913 37.0511 4.5539
If we look at the printed energy decomposition
NSTEP ENERGY RMS GMAX NAME NUMBER
458 -1.0486E+04 3.9749E-01 8.0012E+00 CH3 3672
BOND = 162.6406 ANGLE = 533.3261 DIHED = 1786.8891
VDWAALS = -1863.1683 EEL = -15763.2530 HBOND = 0.0000
1-4 VDW = 758.2414 1-4 EEL = 4176.2565 RESTRAINT = 0.0000
PM3ESCF = -276.8181
we shall notice that the QM/MM result has an extra field (PM3ESCF). This is the energy due to the quantum part of the calculation (the "catalytic triad" + ligand) within the presence of the charge field of the MM part