
When you first start up a program you will see a main display window and the manubar and toolbar across the top of it. Practically all Jamberoo commands are grouped into several main functional categories listed in the menubar.
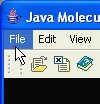
To load a new molecule start a program and select File menu from the menubar
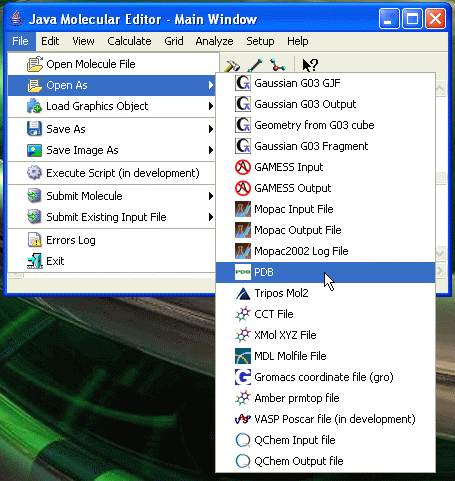
Then select Open as from the File menu and select a molecule file format
A program will open an Open File dialog.
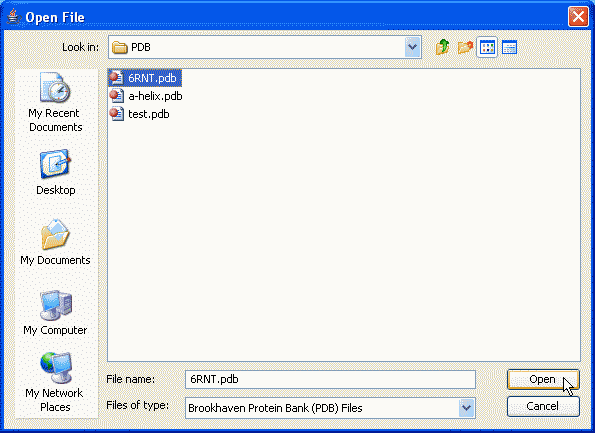
An Open File dialog allows to move through the directory structure and select a file to load. Select a file and click on the Open button
Molecule is displayed in the main window:
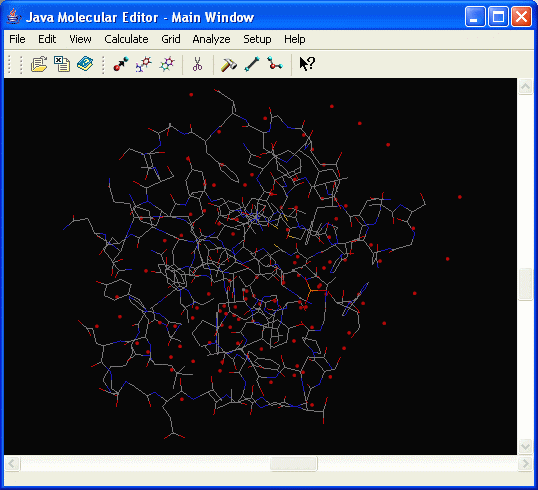
Alternatively, Open File dialog can be activated using the Open File icon on the toolbar:
![]()
All manipulations with molecular structures are accomplished by pressing mouse button and then moving the mouse. It is strongly advisable to use a three button mouse.
Hold down the left mouse button and then move the mouse
Hold down the right mouse button and then move the mouse
Hold down the middle mouse button and then move the mouse
Send all questions, suggestions and comments to Vlad (vvv900@gmail.com)
Dr. Vladislav Vasilyev
Supercomputer Facility,
The Australian National University,
Canberra, ACT, 0200, Australia
[Home] [Applications] [Applets] [API] [E-mail me]